
Vyšetření metodou MLPA - SMA a Connexin 26
Metoda MLPA
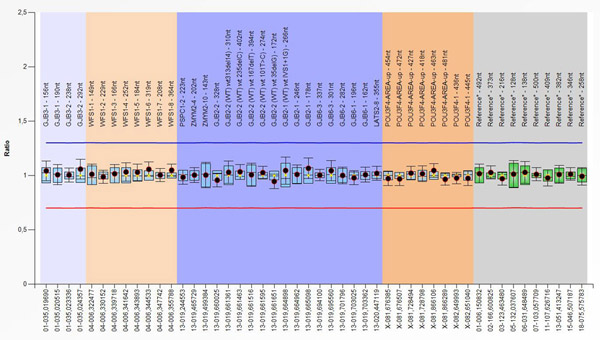
Princip MLPA metody je založen na amplifikaci až 60 sond, z nichž každá detekuje specifickou DNA sekvenci. Vzorek DNA musí obsahovat minimálně 20 ng lidské DNA (asi 3000 buněk). Jedná se tedy o semi-kvantitativní metodu využívanou k vyšetření DNA o délce přibližně 60 nukleotidů během jedné reakce založené na multiplex PCR. MLPA reakce je jedinečná tím, že nejsou amplifikovány cílové sekvence, ale MLPA sondy, které se na tyto cílové sekvence hybridizují.
Na vyšetřované místo se musí navázat dvě specifické DNA sondy, které se uprostřed spojí (ligují). Ligace proběhne jen tehdy, pokud při předchozí hybridizaci specifické sondy nasedly na přilehlé cílové sekvence a mohly tak být následně spojeny do jedné sekvence. Následuje PCR reakce, ve které se všechny ligované sondy namnoží (amplifikují) a následně jsou fragmenty separovány a vizualizovány pomocí kapilární elektroforézy.
Molekulárně genetické vyšetření onemocnění SMA
Základní stavební složkou nervového systému jsou neurony. Neurony se během života nevytváří, ani nejsou schopné obnovy. Pokud se poškodí nebo zahynou, je jejich ztráta pro tělo nevratná. Onemocnění způsobená progresivní degenerací a úhynem neuronů se nazývají neurodegenerativní onemocnění . Projevují se problémy s pohybovým aparátem a zhoršujícími se duševními schopnostmi.
Spinální muskulární atrofie (SMA) je závažné neurodegenerativní onemocnění charakterizované degenerací motorických (pohybových) neuronů v předních rozích míchy. Dle klinických projevů rozlišujeme tři typy SMA - SMA I (nejtěžší forma), II (střední forma) a SMA III (juvenilní forma). Nemocní jedinci trpí různými stupni poruch pohybového aparátu, liší se také rychlost rozvoje onemocnění.
SMA I je nejtěžší forma onemocnění - postižení jedinci se dožívají 2 let věku. SMA II představuje středně těžkou formu onemocnění s nástupem mezi 7. - 18. měsícem života. Častou příčinou úmrtí je respirační selhání v období adolescence. SMA III je tzv. juvenilní forma SMA, první příznaky ochablosti svalstva se projevují po 18. měsíci života, postižené osoby jsou schopné samostatné chůze až do progrese onemocnění a dožívají se dospělosti.
SMA je druhým nejčastějším autozomálně recesivním onemocněním s incidencí 1/10 000 novorozenců a s frekvencí přenašečů až 1/40. SMA je způsobena mutacemi v genu SMN1 (Survival Motor Neuron 1 gene), který se nachází v komplexní oblasti 5q13. V tomto lokusu leží také homologní pseudogen SMN2, jež se liší od genu SMN1 pouze v 5 nukleotidech. Gen SMN2 tvoří jen 10-15% účinného proteinu SMN ve srovnání s genem SMN1. Vzhledem k vysoké homologii obou genů vzácně dochází ke konverzi genu SMN1 do genu SMN2. SMA je v 95 % případů způsobena homozygotní delecí exonu 7 genu SMN1 (v důsledku delece nebo genové konverze SMN1 na SMN2). Přibližně 5 % pacientů je složenými heterozygoty, kdy je delece exonu 7 v SMN1 genu, nebo genová konverze spojena s intragenovou mutací.
Většina přenašečů SMA má pouze jednu kopii exonu 7 SMN1 genu a nejsou postiženi. Delece exonu 7 a 8 v SMN2 genu nemá klinické důsledky, ale vyšší počet kopií exonu 7 a 8 v SMN2 genu u pacientů se spinální muskulární atrofií znamená mírnější projev SMA. Jen asi 60-70 % jedinců má dvě kopie exonů 7 a 8 SMN2.
Molekulárně genetické vyšetření onemocnění dědičné hluchoty a genu Connexin 26
Jeden z možných způsobů klasifikace ztráty sluchu je dělení na syndromové a nesyndromové. Syndromová ztráta sluchu (20-30% dědičných hluchot) znamená pouze jeden z dalších zdravotních projevů celého konkrétního syndromu. Mnohem častěji se však vyskytuje tato vada samostatně. Až 80 % geneticky podmíněných nesyndromových ztrát sluchu se dědí autosomálně recesivně. Ve více než polovině případů autozomálně recesivní ztráty sluchu jsou příčinou mutace v genu GJB2, lokalizovaném na chromozomu 13 (13q12), kódujícím Connexin 26. Connexiny jsou membránové proteiny, které propojují buňky vnitřního ucha a umožňují tak mezibuněčnou komunikaci sluchového aparátu.
Gen pro Connexin 26 je poměrně malý, přesto v něm bylo nalezeno velké množství mutací. V evropské populaci bylo detekováno ve velké míře (40-80 %) především chybění (delece) jednoho nukleotidu (guaninu - G) v pořadí 6 guaninů v pozici 30-35 genu GJB2 (mutace 35delG), které vede k předčasnému ukončení tvorby proteinu.
...CTGGGGGGTGTGAACAAACAC..... normální sekvence = funkční gen = normální sluch
...CTGGGGG...TGTGAACAAACAC..... mutace = nefunkční gen = vada sluchu
V české populaci byla zjištěna frekvence přenašečů 1/49. Rodiče jsou typicky zdraví přenašeči (heterozygoti), pro potomstvo je následně 25% riziko postižení obou forem genu (od matky i otce) a vzniku poruchy sluchu. Rodinný výskyt sledujeme často v manželstvích neslyšících osob nebo u příbuzenských vztahů. U slyšících přenašečů je vhodné vyšetření jejich partnerů, protože frekvence heterozygotů v běžné slyšící populaci je poměrně vysoká (kolem 3 %). Kromě nejfrekventovanější zmíněné mutace 35delG může docházet k dalším méně častým změnám, které také detekujeme pomocí metody MLPA.
Námi používaný diagnostický kit je schopný detekovat i mutace v jiných genech (GJB3, GJB6, POU3F4, WFS1), ovšem jejich podíl na celkovém výskytu poruch sluchu je v naší populaci pouze zanedbatelný.
O laboratoři
Pro pacienty a samoplátce
- Trochu o genetice
- Co je genetické testování ?
- Jak mám rozumět výsledkům?
- Mám mít z odběru na testování obavy?
- Ceník vyšetření
Pro odbornou veřejnost
Součásti institutu
Adresa laboratoře
FertiCare SE
provozovna Karlovy Vary
Laboratoř lékařské genetiky
Bělehradská 1042/14
36001 Karlovy Vary
Telefony:
Objednání: 353 433 958, 776 202 225
Laboratoř: 353 433 964